Computational Recipe for Efficient Description of Large-Scale Conformational Changes in Biomolecular Systems
By Mahmoud Moradi and Emad Tajkhorshid.
Published in J Chem Theory Comput.M 2014 Jul 8;10(7):2866-2880.
PMID: 25018675. PMCID: PMC4089915 [Available on 2015/6/3]. Link to publication page.
Core: Computational Modeling Core.
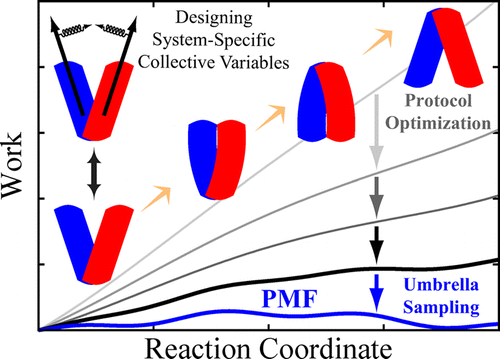
Abstract
Characterizing large-scale structural transitions in biomolecular systems poses major technical challenges to both experimental and computational approaches. On the computational side, efficient sampling of the configuration space along the transition pathway remains the most daunting challenge. Recognizing this issue, we introduce a knowledge-based computational approach toward describing large-scale conformational transitions using (i) nonequilibrium, driven simulations combined with work measurements and (ii) free energy calculations using empirically optimized biasing protocols. The first part is based on designing mechanistically relevant, system-specific reaction coordinates whose usefulness and applicability in inducing the transition of interest are examined using knowledge-based, qualitative assessments along with nonequilirbrium work measurements which provide an empirical framework for optimizing the biasing protocol. The second part employs the optimized biasing protocol resulting from the first part to initiate free energy calculations and characterize the transition quantitatively. Using a biasing protocol fine-tuned to a particular transition not only improves the accuracy of the resulting free energies but also speeds up the convergence. The efficiency of the sampling will be assessed by employing dimensionality reduction techniques to help detect possible flaws and provide potential improvements in the design of the biasing protocol. Structural transition of a membrane transporter will be used as an example to illustrate the workings of the proposed approach.
