Exploring the Conformational Transitions of Biomolecular Systems Using a Simple Two-State Anisotropic Network Model
By Avisek Das, Mert Gur, Mary Hongying Cheng, Sunhwan Jo, Ivet Bahar, and Benoît Roux.
Published in PLoS Computational Biology 10(4):e1003521 on April 3rd, 2014. PMID: 24699246. PMCID: PMC3974643. Link to publication page.
Core Facility: Computational Modeling Core
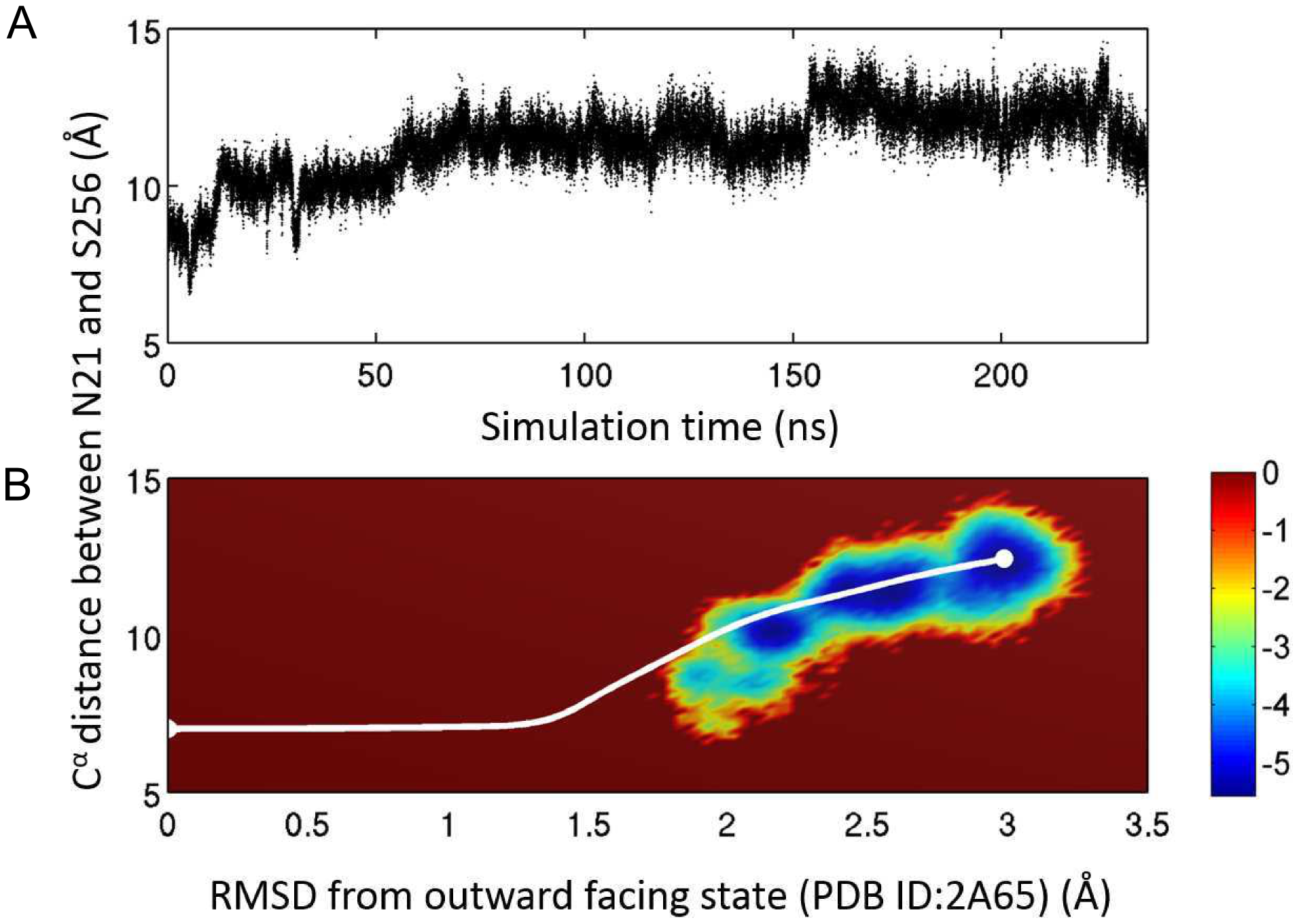
Figure 4. Comparison with all-atom simulation results. A. Time trace of the distance between atoms of residues 21 and 256 from a 235 ns long conventional MD simulation of the fully solvated system. The simulation was started from a structure obtained from a targeted MD simulation originated from the OFc state. The system undergoes a spontaneous transition to the IFo state. B. Comparison of the ANMPathway method and all-atom MD in the space of two order parameters. The all-atom MD results are shown as a pseudo free energy landscape
, where P is the 2D distribution. The color-scale goes from blue (low energy) to red (high energy). The pathway predicted by ANMPathway (white line) goes mostly through the low energy regions of the free energy landscape.
Abstract
Biomolecular conformational transitions are essential to biological functions. Most experimental methods report on the long-lived functional states of biomolecules, but information about the transition pathways between these stable states is generally scarce. Such transitions involve short-lived conformational states that are difficult to detect experimentally. For this reason, computational methods are needed to produce plausible hypothetical transition pathways that can then be probed experimentally. Here we propose a simple and computationally efficient method, called ANMPathway, for constructing a physically reasonable pathway between two endpoints of a conformational transition. We adopt a coarse-grained representation of the protein and construct a two-state potential by combining two elastic network models (ENMs) representative of the experimental structures resolved for the endpoints. The two-state potential has a cusp hypersurface in the configuration space where the energies from both the ENMs are equal. We first search for the minimum energy structure on the cusp hypersurface and then treat it as the transition state. The continuous pathway is subsequently constructed by following the steepest descent energy minimization trajectories starting from the transition state on each side of the cusp hypersurface. Application to several systems of broad biological interest such as adenylate kinase, ATP-driven calcium pump SERCA, leucine transporter and glutamate transporter shows that ANMPathway yields results in good agreement with those from other similar methods and with data obtained from all-atom molecular dynamics simulations, in support of the utility of this simple and efficient approach. Notably the method provides experimentally testable predictions, including the formation of non-native contacts during the transition which we were able to detect in two of the systems we studied. An open-access web server has been created to deliver ANMPathway results.
 and the closed (right, PDB ID: 1AKE) states. The LID and the NMP domains are shown in red and orange respectively. The CORE domain and the rest of the protein are shown in blue. The central structure is the trace of the transition state produced by ANMPathway. B. The energy of the system along the transition. Total number of images in the pathway is 100, RMSD between two consecutive images is ~0.1 Å. The transition state corresponds to image 89. C. Cumulative squared cosines between ANM modes and the change in structure between the initial state and a few selected conformers (images) along the transition pathway. The modes were calculated for the starting structure (open state). Here and in the counterparts generated for other test cases, the lowest frequency (slowest modes) end of the graph is enlarged in the inset. The force constants and cut-offs for both the end-states were set to 0.1 kcal/(mol Angstrom) and 15 Å in all applications in the present study, and no energy offsets were used for either of the end structures.")
 and Open (top, diamond) substates of AK for the crystal structures (black) and their MD equilibrated (magenta) conformers. ANMPathway and three coMD [64] pathways are depicted as black, blue, magenta and green dots, respectively, with the arrows indicating the direction of reconfiguration in each case. The relative positions of the domains are defined similar to Beckstein et al. [72] and Gur et al. [64]. The NMP-CORE angle is the angle between the centers of mass of three segments I90-G100, L115-V125 and L35-A55, based on atoms and the LID-CORE angle is the angle between centers of mass of atoms of L115-V125, I179-E185 and V125-L153.")
 and the inward-facing open (IFo) states of leucine transporter, LeuT. A. Structures of the OFc state (left, PDB ID: 2A65) and the IFo state (right, PDB ID: 3TT3). The scaffold domain, which does not undergo significant conformational changes, is shown in gray and the rest of the protein is shown in blue (EL4), red (TM1a), orange (TM1b), purple (TM2 and TM7), yellow (TM5), green (TM6a) and lime (TM6b). The central structure is the trace of the transition state produced by the ANMPathway method. B. The energy of the system along the transition. Total number of images in the pathway is 95, RMSD between two consecutive images is ~0.05 Å. The transition state corresponds to image 40. C. Cumulative squared cosines between the structural change to reach a few selected conformers/images along the transition pathway and the ANM modes accessible to the starting (outward-facing occluded) structure.")
 state along the computed transition pathway between outward-facing open (OFo) and inward-facing open (IFo) states of LeuT. A. Relative orientations of TM1, TM6 and TM10 of LeuT in the OFo (orange), OFc (yellow) and IFo (cyan) crystal structures. The OFo crystal structure is shown in transparent cartoon. Inward tilting of the TM1b and TM6a segments contributes to the closure of the extracellular vestibule, indicating the decreased distances of TM1b-TM10 and TM6a-TM10. Outward tilting of TM1a dominates the opening of the intracellular vestibule, resulting in the increased distance between TM1a andTM6b. B. Variation of the center of mass (COM) distances of TM1a-TM10 (red+), TM6b-TM10 (blue+), and TM1a-TM6b (gray+) as the LeuT undergoes transition from the OFo to IFo states. For COM distance calculations, TM1a (R11 to A22), TM1b (L25 to A35), TM6a (G242 to L255), TM6b (F259 to Y268), and TM10 (K398 to V412) atoms are taken from ANMPathway calculations of LeuT from IFo to OFo. Yellow arrows point to the values found in the OFc crystal structure. Clearly, occluded intermediates are identified by ANMPathway calculations (highlighted by the horizontal blue arrow).")
 and the z component of the displacement of the center of mass (COM) of the transport domain (based on atoms) with respect to the initial crystal structure. Negative values along the ordinate point to the IC region. The dots of different colors are the projection of conformers sampled in unbiased all-atom MD simulation runs initiated from various points along a TMD path between the two end-states. For the middle section the data clouds from the unbiased MD simulations cluster around the pathway predicted by ANMPathway.")
