Rapid parameterization of small molecules using the Force Field Toolkit
By Christopher G. Mayne, Jan Saam, Klaus Schulten, Emad Tajkhorshid, and James C. Gumbart.
Published in Journal of Computational Chemistry, 34(32):2757-2770 on December 15, 2013. PMID: 24000174. PMCID: PMC3874408. Link to publication page.
Core Facility: Computational Modeling
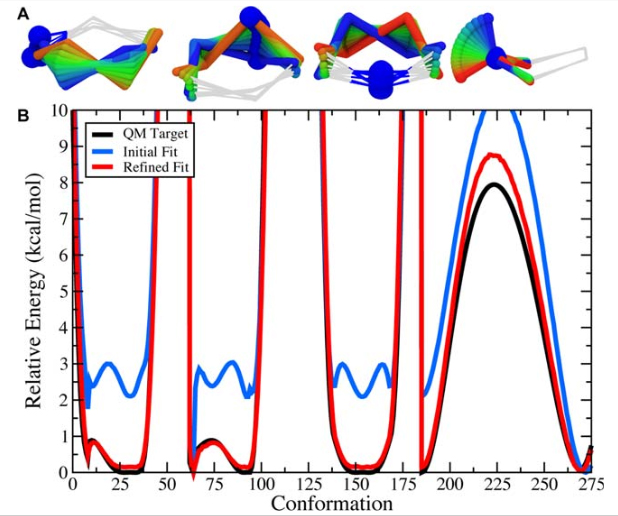
Figure 9. A) The scanned torsions for pyrrolidine were loaded into VMD and colored by iteration (blue to red) to provide a structural context for assessing the dihedral PESs. B) The cyclic structure of pyrrolidine yields a complex dihedral scan profile, in which much of the fine detail lies below 1 kcal/mol; however, an accessible barrier of 8 kcal/mol exists for the N-inversion. The refined MM parameters yield a PES (red) that reproduces the QM PES (black) with excellent agreement.
Abstract
The inability to rapidly generate accurate and robust parameters for novel chemical matter continues to severely limit the application of molecular dynamics simulations to many biological systems of interest, especially in fields such as drug discovery. Although the release of generalized versions of common classical force fields, for example, General Amber Force Field and CHARMM General Force Field, have posited guidelines for parameterization of small molecules, many technical challenges remain that have hampered their wide-scale extension. The Force Field Toolkit (ffTK), described herein, minimizes common barriers to ligand parameterization through algorithm and method development, automation of tedious and error-prone tasks, and graphical user interface design. Distributed as a VMD plugin, ffTK facilitates the traversal of a clear and organized workflow resulting in a complete set of CHARMM-compatible parameters. A variety of tools are provided to generate quantum mechanical target data, setup multidimensional optimization routines, and analyze parameter performance. Parameters developed for a small test set of molecules using ffTK were comparable to existing CGenFF parameters in their ability to reproduce experimentally measured values for pure-solvent properties (<15% error from experiment) and free energy of solvation (±0.5 kcal/mol from experiment).
This publication was the Cover Article for the Journal of Computation Chemistry, Volume 34, Issue 32:

 Each water interaction site (blue spheres) is identified as a hydrogen bond “acceptor” or “donor” by the orientation of the water molecule with respect to the interaction site. B) The position of the interacting atom of the water molecule (H or O; red spheres) is determined based on the geometry of the interaction site to reduce steric interactions with covalently bound neighbors (grey spheres).")
 Scanned dihedrals are auto-detected from a parameter file, or specified manually in the GUI. B) When a specific entry is selected, the corresponding dihedral is highlighted in the main VMD window. C) ffTK employs a bidirectional scanning technique that scans the energy regime relevant to parameterization and avoids complications of starting the scan from high-energy conformations. D) The result of the scan can be directly loaded into VMD for visual inspection (colored by step proceeding from blue to red).")
 The COLP extracts relevant data from the charge optimization log file for visual analysis. These data include the total objective function, the individual contributions from energy, distance, and dipole moment to the objective function (shown), as well as, the minimum interaction energy and distance for each interaction site probed during the optimization. (Bottom) During dihedral fitting, the MM PES computed for each refinement step can be visualized using an embedded plotting utility for comparison against the QM target PES. The data from both plotting utilities can be directly exported to file for import into popular plotting applications to generate publication-quality plots.")

 level of theory. The calculations were run in Gaussian; however, all input files were generated from ffTK and the results visualized in VMD.")
