Structural Refinement of Proteins by Restrained Molecular Dynamics Simulations with Non-interacting Molecular Fragments
By Rong Shen, Wei Han, Giacomo Fiorin, Shahidul M. Islam, Klaus Schulten, and Benoît Roux.
Published in PLoS Comput Biol. 2015 Oct 27;11(10):e1004368. PMID: 26505197. Link to Pubmed page.
Core Facility: Computational Modeling.
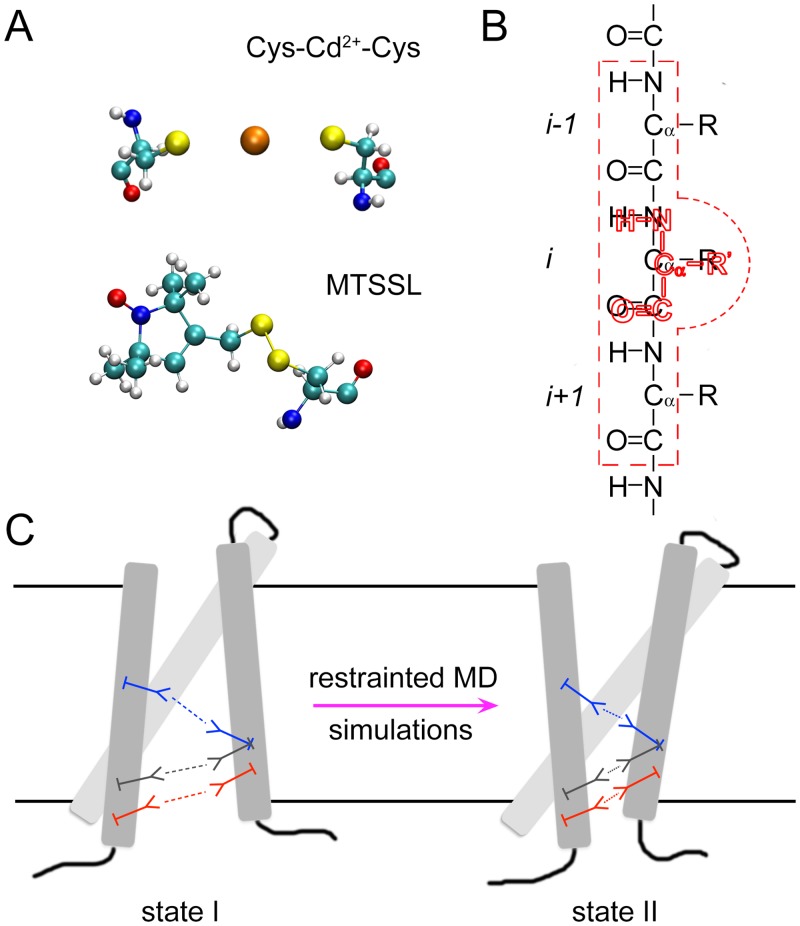
Figure 1, Schematic of the molecular fragments method. (A) Each structural constraint (e.g., a metal ion bridge or a spin-label) is present as a molecular fragment with all atomic details in the system. (B) The residue(s) in the molecular fragment (colored in red) is/are attached to the targeting residue(s) in the wild-type protein, via harmonic restraints, with their backbone atoms (N, C, O and Cα) staying on top of each other, respectively. The residue in the molecular fragment does not have interactions with its targeting residue in the wild-type protein (residue i), and the interactions between the backbone atoms of the residue in the molecular fragment and the backbone atoms of the two nearby residues in the wild-type protein (residues i-1 and i+1; embraced by red dashed lines) are also turned off. (C) During the restrained MD simulations, the interactions within each molecular fragment are evaluated accurately triggering the conformational changes of the wild-type protein, while different molecular fragments do not have interactions with one another.
Abstract
The knowledge of multiple conformational states is a prerequisite to understand the function of membrane transport proteins. Unfortunately, the determination of detailed atomic structures for all these functionally important conformational states with conventional high-resolution approaches is often difficult and unsuccessful. In some cases, biophysical and biochemical approaches can provide important complementary structural information that can be exploited with the help of advanced computational methods to derive structural models of specific conformational states. In particular, functional and spectroscopic measurements in combination with site-directed mutations constitute one important source of information to obtain these mixed-resolution structural models. A very common problem with this strategy, however, is the difficulty to simultaneously integrate all the information from multiple independent experiments involving different mutations or chemical labels to derive a unique structural model consistent with the data. To resolve this issue, a novel restrained molecular dynamics structural refinement method is developed to simultaneously incorporate multiple experimentally determined constraints (e.g., engineered metal bridges or spin-labels), each treated as an individual molecular fragment with all atomic details. The internal structure of each of the molecular fragments is treated realistically, while there is no interaction between different molecular fragments to avoid unphysical steric clashes. The information from all the molecular fragments is exploited simultaneously to constrain the backbone to refine a three-dimensional model of the conformational state of the protein. The method is illustrated by refining the structure of the voltage-sensing domain (VSD) of the Kv1.2 potassium channel in the resting state and by exploring the distance histograms between spin-labels attached to T4 lysozyme. The resulting VSD structures are in good agreement with the consensus model of the resting state VSD and the spin-spin distance histograms from ESR/DEER experiments on T4 lysozyme are accurately reproduced.
Author Summary
Knowledge of multiple conformational states of membrane transport proteins is a prerequisite to understand their function. However, the determination of atomic structures for all these states with conventional high-resolution approaches can be very challenging due to inherent difficulties in high yield purification of functional membrane transport proteins. Various complementary structural information of proteins in their native states can be obtained by a variety of biophysical and biochemical methods with site-directed mutations. Here, a novel restrained molecular dynamics structural refinement method is developed to help derive a structural model that is consistent with experimental data by incorporating all the experimental constraints simultaneously through the use of non-interacting all-atom molecular fragments. The method can be easily and effectively extended to incorporate many kinds of structural constraints from a variety of biophysical and biochemical experiments, and should be very useful in generating and refining models of proteins in specific functional states.
